- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Dear Community;
We usually read that parallel computation (either OpenMP or MPI) is not deterministic. This is usually easy to visualize in I/O. However, I am comparing my Serial and Parallel version (MPI) of the same code and the difference is close to 0.3% in the error. The error is defined as the maximum difference between two subsequent time steps. Thus, the difference between the error of the serial version and the MPI is close to 0.3 %. Although the error is small, the solution presents slight differences, 1% in certain regions of the computation. When I increase the number of processes then that difference is more notorious. Again, that difference is still small.
I was wondering about the cause of this problem. Also, I would like to ask two more questions. I was thinking that maybe the problem comes because I am using -O3 flags. I have heard that it is usually not recommended to use optimization with MPI... but I have not found anything consistent to back up my decision.
As far as good practice, can we change MPI_REDUCE + MPI_BROADCAST by just a MPI_ALLREDUCE ? Again, I have heard that it is not good to change those for one MPI_ALLREDUCE, but it seems that the literature does not prove it directly. I am using the MPI_REDUCE + MPI_BROADCAST to use the same time step in the whole computation.
Finally, as far as 2D and 1D domain decomposition is there any rule that allows to decide between these two approaches? My problem is 2D but I have seen other people using 2D decomposition and they end up with ghost cells in four directions, i.e: Up, down, left and right. I only have left and right neighbours.
Thanks you all!!!
- Tags:
- Parallel Computing
Link Copied
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
>>We usually read that parallel computation (either OpenMP or MPI) is not deterministic
This statement has to be qualified.
When a serial program produce results containing error (lack of precision thus containing round off errors), then partitioning the problem will also produce results containing error (lack of precision thus containing round off errors). When these errors are accumulated, either within the serial application. or in pieces in a parallel process, then the order of accumulation can affect the results. IOW results typically differ. Note, in the serial program, if you perform the accumulation in the other direction (e.g. indexing an array of partial results backwards), you can end up with different values.
It should not be overlooked that when partitioning a problem (parallelizing), that the partitioned program, under the right circumstances, will produce more accurate results. IOW your 0.3% could potentially be 0.3% closer to the correct result.
Explanation: When accumulating results of relatively large values with relatively small values (with long sequence of binary fractional values), that the small values will have a larger portion of the fractional value, and in some cases a goodly portion of the integral part discarded. When partitioning the problem a lessor amount of this loss tends to occur.
The real questions for you to answer are:
Can you figure out why the difference?
And more importantly, is the difference in the direction of a better result?
Here is a suggested test. If your serial program produces an array of results of which you reduce to a sum, then sort the array by magnitude of absolute values and produce the sum in order of smallest magnitude (not absolute), and compare the results. I would expect the difference to be larger than 0.3%. If not, then your conversion to MPI may have an underlying problem.
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
At the heart of it all is the fact that floating-point arithmetic is not associative; for example, a + (b + c) is not equal to (a + b) + c. It follows that a parallel reduction produces a different result than a serial reduction, even when the parallel reduction is deterministic. A full description can be found in a textbook on Numerical Analysis such as chapter one of Atkinson. It is a common mistake to believe a serial result is more accurate a parallel result.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Thank you very much for your kind replies. Here I want to make a disclaimer, I am a mechanical engineer with very but very few knowledge on compilers, computers architecture an so forth. I sued computers to solve my problems, although I know MPI,OpenMP and FORTRAN very well; but I will not call my self as an expert in this field. Therefore, somethings may be very challenging for me to get. Nonetheless, thank you very much for your time in this threads. I still have few questions from the previous replies.
Mr. Greeg S.. I guess that the associative properties does not hold for parallel computing since the round off error behaves differently in each process, since each process have a different chunk of the domain. However, I would like to know why you said that we usually believe that a serial results is more accurate than the parallel one. Finally, you said that under the right conditions partioning the problem will deliver more accurate results, why is that? if again, the ghost cell carry all that information to the neighbouring cells. What happens at the compiler level or hardware level that improves the solution.
Any suggestions with the MPI_REDUCE = MPI_BROADCAST changed by MPI_ALLREDUCE ? and -O3 for MPI?
thanks you all!
Mr. jimdempseyatthecov
I see the difference of the sum in different direction, it makes a lot of sense since the accumulative error due to the difference direction will be different. However, when we send the ghost cells to the other neighbours, that problem may be suppress because the ghost cells may contain that "accumulative error" implicit in it.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Floating-point arithmetic is not associative, period. Regardless of parallelism.
For example, adding 0.1 + (0.2 + 0.3) may yield 0.59999999999999998, and adding (0.1 + 0.2) + 0.3 may yield 6.0000000000000009. Both results are correct for floating-point arithmetic. Neither is a problem. I encourage you to read a full discussion such as chapter one of Atkinson, because one cannot effectively compress this topic into a brief explanation.
Differences between serial and MPI parallel results are not a problem, they are normal. When a reduction happens in parallel, the order of the floating-point operations is changed. For a new order of operations, the accumulated error may be larger or smaller.
Avoiding -O3 with MPI sounds like bad advice.
I do not know any reason to choose reduce + broadcast over allreduce.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
>> I would like to know why you said that we usually believe that a serial results is more accurate than the parallel one.
This is (I assume) due to the serial program being used to produce the reference dataset. And then assuming any variation from this is to be considered an error. Even though the deviating program may in fact be producing closer results.
>>under the right conditions partioning the problem will deliver more accurate results, why is that?
You are aware that floating point operations can produce round off errors. While the round off error of a single operation can be thought of 1/2 lsb in the result, what is actually happening for addition and subtraction is one of the arguments of the operation has no loss, while a second argument has zero to all bits of loss. The larger the difference in magnitude, the more bits are lost. For summation, the closest result is produced by summing from least magnitude to largest magnitude, though this may be impractical to sort the array of values to be summed.
Assume as an example, you have a 2 digit calculator. Which can produce non-zero results:
0.1, 0.2, ... 9.9, 10., 11., ... 99.
What happens when you sum the numbers:
10, 1.0, 1.9, 1.8
The answer you get is 13
sum the numbers in reverse order (lowest to highest)
The answer is 14
Which is closer to the correct answer of 14.7?
Assume you parallelize the first instance
10 + 1.0 || 1.9 + 1.8
11 || 3.7
reduce
14
(the above was done without round-up for illustrative purposes)
In the above "parallel" example, the end result was closer. Different orders of numbers will effect the result.
RE: ghost cells
There is insufficient information as to how these cells are produced and used. In my experience, the boundary cells of a domain can be representative of the corresponding boundary of the adjacent domain (tile). These numbers can be synchronized at each iteration of a simulation .OR. to reduce synchronization they can be extrapolated for a few iterations and synchronized/resolved less frequently.
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Mr. Dempesey;
I greatly appreciate your kindness and time in this question. Your explanation helped me a lot.
Thanks
Julio
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Dear Mr. Dempesey;
I have checked my results under different conditions, and I found the same trend. The solution varies when I increase the number of npes. If I keep the npes small, i.e :2,3,4; the solution approaches the Serial Version (Here I am assuming the Serial is right).
I was discussing this with my advisor, and We both agreed that the round-off error in fortran is fixed since we specified each variables as kind=8. Therefore, we do not see how the floating point operation affects the round-off error.
Based on your last explanation, I concluded that parallel computing yields better results because reducing the floating point the round-off error decreases too. However, how is that possible if the we are using the same precision in each npes.
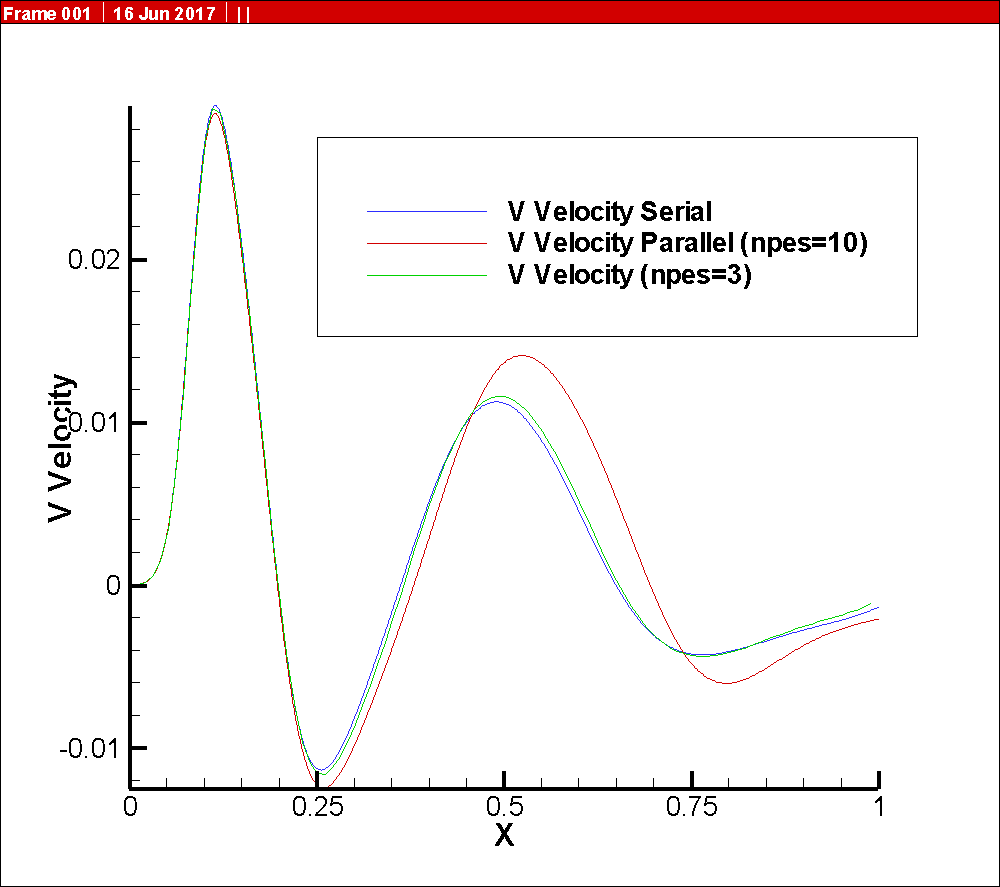
I am attaching the solution, that is the vertical component of the velocity vector at some point in the channel. It is interesting to note that at the first part of the solution the three solution are very similar, but the differences start after the first quarters (approximate). The difference is around 32% (between the serial and the one with npes=10).
It would be very interesting knowing your point of view.
Thanks in advance
{kind=link}
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Even double-precision floating-point has round-off error and is not associative. The perturbations in parallel reductions are well known and well documented, see for example http://bebop.cs.berkeley.edu/reproblas/docs/talks/SIAM_AN13.pdf
Both you and your advisor would do well to hasten to read chapter one of Atkinson with alacrity. It would be much easier and more comprehensive than trying to teach the material question-by-question in this forum.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Thank you Mr. Greeg, my ultimate goal was to bother the people of this forum.
I am quite sure, my advisor understands very well errors and their sources. I already read the first chapter and it does not address my specific problem, Why the solution is different when the number of CPUS increases (Difference 32 %)? Why is this difference when the number of CPUS (up to 3) is very small (difference 0.3%). This problem is not addressed in the textbook, and I have not found anything similar on the web. You are stressing to much in the reduction but that reduction is only for reducing the error and time step (both are used to stop the iterations, but they do not control the final stage). Because, delta T is the same in all the ranks.
Finally, I strongly believe that forums are for these type of healthy discussions. I am not asking for solving my problem, just different perspectives and advice.
Thanks for the material you recommended.
Julio
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
The variation you are seeing does not look especially large. Validated, provably correct industry codes produce much worse examples. Some applications add a flag or command which allows the user to select serial reductions, sacrificing performance for consistency.
This is what happens with floating-point arithmetic when the order of operations is changed, and it gives an idea how much error +/- is in the serial calculation. The same kind of variation can occur moving from one architecture to another as well, both serial. Some steps can be taken to reduce the effect, by ordering operations to avoid catastrophic cancellation and such.
If you don't like the normal explanation about order of floating-point operations, the alternative would be a bug in your parallel code. The simple act of calculating on multiple processors does not produce different results.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Thank you Mr. Gregg. I have also thought about a bug, but for small number of CPUS the difference is negligible (0.5%). As a result, I believed that the implementation is right.
I have also checked thoroughly the behavior of the code when increasing the CPUS, checking he ghost cells in each arrays. For these checks everything looks perfect. Nonetheless, I am not saying the implementation is 100% free of bugs. I will try to setup other tests and see how it behaves.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
>> checking (t)he ghost cells in each arrays
Can you show how you implement your ghosting cells (including time step integration).
Note, your serial implementation has no ghosting cells.
3-way parallel has two ghosting cell boundaries
10-way parallel has nine ghosting cell boundaries.
The skew observed in your data leads me to suspect that your ghosting method is the cause as opposed to round off error.
One of the characteristics of introducing inter-process coordination data points (aka ghosting cells), is this can introduce a time delay skew in results data.
Are you using Euler method for integration or one of the Adams multi-step methods for integration?
Euler tends to accumulate errors more than Adams (YMMV).
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Hi Mr. Jim thanks for your advise and your concern in this issue. I will share part of my implementation.
The following section takes care of the communication, since I am using persistent communication.
**********************************************************************************************
Subroutine MPI_StartUp
use Variables
use mpi
implicit none
!Starting up MPI
call MPI_INIT(ierr)
call MPI_COMM_SIZE(MPI_COMM_WORLD,npes,ierr)
call MPI_COMM_RANK(MPI_COMM_WORLD,MyRank,ierr)
!Compute the size of local block (1D Decomposition)
Jmax = JmaxGlobal
Imax = ImaxGlobal/npes
if (MyRank.lt.(ImaxGlobal - npes*Imax)) then
Imax = Imax + 1
end if
if (MyRank.ne.0.and.MyRank.ne.(npes-1)) then
Imax = Imax + 2
Else
Imax = Imax + 1
endif
! Computing neighboars
if (MyRank.eq.0) then
Left = MPI_PROC_NULL
else
Left = MyRank - 1
end if
if (MyRank.eq.(npes -1)) then
Right = MPI_PROC_NULL
else
Right = MyRank + 1
end if
! Initializing the Arrays in each processor, according to the number of local nodes
Call InitializeArrays
!Creating the Data type to care of non contiguous data arrays.
Call MPI_TYPE_VECTOR(Jmax,1,Imax,MPI_REAL8,GhostCells,ierr)
Call MPI_TYPE_COMMIT(GhostCells,ierr)
!This section defines the displacement and communicates the Local Imax among the ranks
! for communications purposes (Valid for BC and I/O)
Displacement = 0
ImaxRecv = 0
if (MyRank ==0) then
do i = 1, npes -1
Displacement(i+1) = ((ImaxRecv(i) - 1) + Displacement(i))-1
end do
end if
Call MPI_GATHER(Imax,1,MPI_INTEGER,ImaxRecv,1,MPI_INTEGER,0,MPI_COMM_WORLD,ierr)
!Creating the channel of communication for this computation,
!Sending and receiving the u_old (Ghost cells)
Call MPI_SEND_INIT(u_old(2,1),1,GhostCells,Left,Tag,MPI_COMM_WORLD,Req(1),ierr)
Call MPI_RECV_INIT(u_old(Imax,1),1,GhostCells,Right,Tag,MPI_COMM_WORLD,Req(2),ierr)
Call MPI_SEND_INIT(u_old(Imax-1,1),1,GhostCells,Right,Tag,MPI_COMM_WORLD,Req(3),ierr)
Call MPI_RECV_INIT(u_old(1,1),1,GhostCells,Left,Tag,MPI_COMM_WORLD,Req(4),ierr)
! I have more of these for each variable.
****************************************************************************************************************************
This section is the main program, where I use MPI_REDUCE and MPI_BROADCAST to use the lowest error and time step.
!Setting up the partition for each processor and initializing MPI
Call MPI_StartUp
UpdateBoundaries = 10 !It sets the frecuency for updating the GhostCells
Call Freestream
iRestart = 0
if(iRestart.eq.0)then
Call GridBC
Call Grid
Call WAG
Call BC_Supersonic
else
Call GridBC
Call Grid
Call Read_SolutionRestart
endif
Call MPI_BARRIER(MPI_COMM_WORLD,ierr)
DO kk=1, 2001
!Different subroutines computing stresses and so forth.
! communicating the maximum error among the processes and delta t
Call MPI_REDUCE(eps,epsGlobal,1,MPI_REAL8,MPI_MAX,0,MPI_COMM_WORLD,ierr)
Call MPI_BCAST(epsGlobal,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr)
Call MPI_REDUCE(delta_t,delta_tGlobal,1,MPI_REAL8,MPI_MIN,0,MPI_COMM_WORLD,ierr)
if(MyRank.eq.0) delta_t = delta_tGlobal
Call MPI_BCAST(delta_t,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr)
! Printing the maximum error and the iterations
if(MyRank.eq.0) then
write(*,*) kk,epsGlobal,(kk*delta_t)
write(32,*) kk,epsGlobal
endif
Call Swap !This subroutine update the new variables to be used in the next iteration
!Starts the communication
if (mod(kk,UpdateBoundaries).eq.0) then
Call MPI_STARTALL(28,Req,ierr)
Call MPI_WAITALL(28,Req,Status,ierr)
end if
if (epsGlobal.le.10.0**(-8.0)) then
exit
endif
Call MPI_BARRIER(MPI_COMM_WORLD,ierr) ! Barrier in MPI
Enddo
!Output sections
***************************************************************************************************************************
I am using Euler for my time integration and since I am using 1D decomposition and my problem is 2D I only have two ghost cells, one in each direction (horizontal direction)
Thanks in advance.
Julio
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
It appears that you are re-computing a variable called delta_t. Are you using variable time steps?
If so, it may be enlightening to produce and save the table of variable time steps as used by the serial program, and then re-use those time steps in the 3-rank and 10-rank programs. Note, I am not suggesting your final application does this, rather this may isolate where the difference is being generated. Knowing this, you can then devise a workable solution.
I do not see how you are using your ghost cells.
What happens to the results data when you set UpdateBoundaries to every iteration (unknown as to if this is 0 or 1)?
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Hi Jim, thank you for you suggestion. In fact the time step is exactly the same. I printed the Time step from the serial version and the parallel versions under different npes, and the solution is very encouraging because the time step predicted was the same. Well almost the same, a difference of 0.0001%. However, that was not the same for the error. Here I want to draw your attention. I believe, that the error is being reduced and broadcasting out of time or I might be using the errors from the previous time step.
The main code computes the time step almost at the top and the error almost at the end of the main loop. Therefore, I am thinking that when the code hits the MPI_REDUCE and MPI_BROADCAST for the time step, the buffer is ready with the right values, but not the case for the error. Let me show you.
DO kk=1, 2001 Call (name of Subroutine) Call (name of Subroutine) Call Time_Step ! Here is where I compute the time step Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call Update ! Here is where I compute the error ! communicating the maximum error among the processes and delta t Call MPI_REDUCE(eps,epsGlobal,1,MPI_REAL8,MPI_MAX,0,MPI_COMM_WORLD,ierr) Call MPI_BCAST(epsGlobal,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr) Call MPI_REDUCE(delta_t,delta_tGlobal,1,MPI_REAL8,MPI_MIN,0,MPI_COMM_WORLD,ierr) if(MyRank.eq.0) delta_t = delta_tGlobal Call MPI_BCAST(delta_t,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr) ! Printing the maximum error and the iterations if(MyRank.eq.0) then write(*,*) kk,epsGlobal,(kk*delta_t) write(32,*) kk,epsGlobal endif Call Swap !Starts the communication if (mod(kk,UpdateBoundaries).eq.0) then Call MPI_STARTALL(28,Req,ierr) Call MPI_WAITALL(28,Req,Status,ierr) end if if (epsGlobal.le.10.0**(-8.0)) then exit endif Call MPI_BARRIER(MPI_COMM_WORLD,ierr) ! Barrier in MPI Enddo
For restrictions reasons I used (name of Subroutine) instead of the real names, sorry about that. If you see, the subroutine UPDATE might not be finished when I am reducing error. However, some ranks might be lagged behind, I do not know. I have read that the collective communications have implicit barriers, but I am not that sure. I do not what the specifications says. I am thinking about doing the following modifications:
DO kk=1, 2001 Call (name of Subroutine) Call (name of Subroutine) Call Time_Step ! Here is where I compute the time step Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call (name of Subroutine) Call Update ! Here is where I compute the error Call Swap !Starts the communication if (mod(kk,UpdateBoundaries).eq.0) then Call MPI_STARTALL(28,Req,ierr) Call MPI_WAITALL(28,Req,Status,ierr) end if ! Here I will wait until everything is done and every rank has sent/recevied the ghost cells ! updated by Swap. Thus every rank must hit the MPI_WAITALL, I kind of barrier ! communicating the maximum error among the processes and delta t Call MPI_REDUCE(eps,epsGlobal,1,MPI_REAL8,MPI_MAX,0,MPI_COMM_WORLD,ierr) Call MPI_BCAST(epsGlobal,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr) Call MPI_REDUCE(delta_t,delta_tGlobal,1,MPI_REAL8,MPI_MIN,0,MPI_COMM_WORLD,ierr) if(MyRank.eq.0) delta_t = delta_tGlobal Call MPI_BCAST(delta_t,1,MPI_REAL8,0,MPI_COMM_WORLD,ierr) ! Printing the maximum error and the iterations if(MyRank.eq.0) then write(*,*) kk,epsGlobal,(kk*delta_t) write(32,*) kk,epsGlobal endif if (epsGlobal.le.10.0**(-8.0)) then exit endif Call MPI_BARRIER(MPI_COMM_WORLD,ierr) ! Barrier in MPI Enddo
In this way the MPI_WAITALL acts as a barrier, but still I am not sure if thise will require another MPI_BARRIER.
Thanks for your time !!!
Julio
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
!Creating the channel of communication for this computation, !Sending and receiving the u_old (Ghost cells) Call MPI_SEND_INIT(u_old(2,1),1,GhostCells,Left,Tag,MPI_COMM_WORLD,Req(1),ierr) Call MPI_RECV_INIT(u_old(Imax,1),1,GhostCells,Right,Tag,MPI_COMM_WORLD,Req(2),ierr) Call MPI_SEND_INIT(u_old(Imax-1,1),1,GhostCells,Right,Tag,MPI_COMM_WORLD,Req(3),ierr) Call MPI_RECV_INIT(u_old(1,1),1,GhostCells,Left,Tag,MPI_COMM_WORLD,Req(4),ierr)
This looks like you have miss-constructed your ghost cells.
In Fortran, adjacent left-most indexes are adjacent in memory
A(n,m) and A(n+1,m) are in adjacent memory locations.
From my little understanding of what you are trying to do, I think you should be partitioning the second index by the number of ranks.
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
HI Jim, you are right. The problem is that the person who developed the code used fortran but he was thinking in C++. As a results, I needed to create a data type in order to get the right values since they were non-contiguous.
Call MPI_TYPE_VECTOR(Jmax,1,Imax,MPI_REAL8,GhostCells,ierr) Call MPI_TYPE_COMMIT(GhostCells,ierr)
I will keep you posted abut this thread, instead of bugging you all constantly.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
It would be much faster to copy the data to a contiguous buffer before sending.
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
HPC == High Performance Computing
.not.
HPC == High Power Consumption
The data should be laid out, and accessed, in a manner that yields high performance. This may require a little re-conditioning of your C++ programmer for use of Fortran.
Julio, even if you use a strided type, what you are expressing still doesn't make sense.
expressively you have a virtual array A(ImaxGlobal, JmaxGlobal) and you are determining the portion of ImaxGlobal for each rank, Different ranks may have different values for Imax. Each rank then allocating a slice Aslice(Imax, JmaxGlobal). Is this correct??
If this be the case, then
rank 0 has no left ghost cells, and has Aslice(Imax, 1:JmaxGlobal) for right ghost cells
rank 1 has Aslice(1, 1:JmaxGlobal) for left ghost cells, and has Aslice(Imax, 1:JmaxGlobal) for right ghost cells
rank last has Aslice(1, 1:JmaxGlobal) for left ghost cells, and has no right ghost cells
Note, each rank same named Aslice and Imax but have different values and storage locations.
Now then, Assuming (incorrectly for abstraction purposed) Fortran indexed the way C++ does.
Explain why your 2nd MPI_SEND_INIT uses u_old(Imax-1,1) instead of u_old(1, 1)???
*** Note, this is not to say to correct your code that you use u_old(1,1) as that fixes the abstraction of the inverted indexing. The actual fix will involve more than this.
I agree with Greg, in that in cases where you have to use strided array sections that you should copy the stride values into a contiguous temp buffer, and then send that. However, now you visibly see the overhead you have created in the code (an embarrassment in optimization is now exposed).
Jim Dempsey
- Mark as New
- Bookmark
- Subscribe
- Mute
- Subscribe to RSS Feed
- Permalink
- Report Inappropriate Content
Hi Jim, I strongly appreciate your comments. They are very valuable for me.
Here I have two important things to mention. Firstly and more important, I am mechanical engineer with no background on computer sciences. Therefore, my knowledge in memory layout, access and so fort is very limited. Nonetheless, I try to understand as much as possible. Secondly, and more important. My code was develop by my advisor and a former PhD student several years ago, and he said "Julio do not change anything from the Kernel". Although I took MPI classes and few workshops with Jhon Urbanic, still there are lot of things that I need to learn.
Having said that I will reply to your comments.
expressively you have a virtual array A(ImaxGlobal, JmaxGlobal) and you are determining the portion of ImaxGlobal for each rank, Different ranks may have different values for Imax. Each rank then allocating a slice Aslice(Imax, JmaxGlobal). Is this correct??
That's correct. That is why I am using dynamic arrays, because each rank has its own size. Since I am using 1D Decomposition only the 'i' direction is shared among the Ranks available.
If this be the case, then
rank 0 has no left ghost cells, and has Aslice(Imax, 1:JmaxGlobal) for right ghost cells
rank 1 has Aslice(1, 1:JmaxGlobal) for left ghost cells, and has Aslice(Imax, 1:JmaxGlobal) for right ghost cells
rank last has Aslice(1, 1:JmaxGlobal) for left ghost cells, and has no right ghost cells
You are also right here, but my implementation takes care of this issue by itself using MPI_PROC_NULL, look at this:
! Computing neighboars if (MyRank.eq.0) then Left = MPI_PROC_NULL else Left = MyRank - 1 end if if (MyRank.eq.(npes -1)) then Right = MPI_PROC_NULL else Right = MyRank + 1 end if
Now then, Assuming (incorrectly for abstraction purposed) Fortran indexed the way C++ does.
Explain why your 2nd MPI_SEND_INIT uses u_old(Imax-1,1) instead of u_old(1, 1)???
Here you point is also valid, but if you look at the destination it says "right". Thus, if my rank is (npes-1) then the previous snippet takes care.
I agree with Greg, in that in cases where you have to use strided array sections that you should copy the stride values into a contiguous temp buffer, and then send that. However, now you visibly see the overhead you have created in the code (an embarrassment in optimization is now exposed).
Here is the problem. By copying the buffer to a contiguous buffer I will need to create new arrays and one sub-step between the sending and computing. I am still fetching the data in a non-contiguous arrays that will take time, so adding a copy to a contiguous buffer will not relief the non-contiguous fetch. Creating more arrays for this problem will cause more problems because I will need to allocate more variables
My objective was: Create an MPI application in a easy and friendly manner, because this is the philosophy from my advisor and myself. The idea is to share this code with junior members in our team. Still, I want to make sure my implementation is as efficient as possible without being to fancy that only myself will be able to understand the implementation.
- Subscribe to RSS Feed
- Mark Topic as New
- Mark Topic as Read
- Float this Topic for Current User
- Bookmark
- Subscribe
- Printer Friendly Page